
[ad_1]
With the launch of platforms like DALL-E 2 and Midjourney, diffusion generative styles have achieved mainstream recognition, owing to their potential to generate a series of absurd, breathtaking, and typically meme-deserving photos from textual content prompts like “teddy bears doing the job on new AI investigate on the moon in the 1980s.” But a staff of scientists at MIT’s Abdul Latif Jameel Clinic for Device Studying in Overall health (Jameel Clinic) thinks there could be far more to diffusion generative models than just making surreal images — they could speed up the improvement of new medications and lower the likelihood of adverse aspect results.
A paper introducing this new molecular docking model, referred to as DiffDock, will be presented at the 11th International Meeting on Mastering Representations. The model’s unique approach to computational drug style and design is a paradigm change from existing state-of-the-artwork tools that most pharmaceutical firms use, presenting a main prospect for an overhaul of the conventional drug development pipeline.
Medicines normally operate by interacting with the proteins that make up our bodies, or proteins of germs and viruses. Molecular docking was formulated to get insight into these interactions by predicting the atomic 3D coordinates with which a ligand (i.e., drug molecule) and protein could bind together.
Whilst molecular docking has led to the profitable identification of drugs that now take care of HIV and cancer, with each drug averaging a decade of improvement time and 90 % of drug candidates failing costly clinical trials (most scientific tests estimate average drug improvement charges to be all around $1 billion to above $2 billion for every drug), it’s no question that researchers are looking for faster, far more efficient techniques to sift through possible drug molecules.
At this time, most molecular docking equipment applied for in-silico drug structure just take a “sampling and scoring” strategy, searching for a ligand “pose” that finest fits the protein pocket. This time-consuming method evaluates a substantial amount of different poses, then scores them based on how properly the ligand binds to the protein.
In previous deep-learning answers, molecular docking is handled as a regression difficulty. In other phrases, “it assumes that you have a one goal that you’re striving to enhance for and there is a single ideal respond to,” suggests Gabriele Corso, co-creator and next-year MIT PhD university student in electrical engineering and laptop science who is an affiliate of the MIT Computer Sciences and Artificial Intelligence Laboratory (CSAIL). “With generative modeling, you presume that there is a distribution of achievable responses — this is significant in the presence of uncertainty.”
“Instead of a one prediction as formerly, you now enable multiple poses to be predicted, and each a single with a distinctive probability,” adds Hannes Stärk, co-writer and very first-yr MIT PhD scholar in electrical engineering and computer science who is an affiliate of the MIT Personal computer Sciences and Artificial Intelligence Laboratory (CSAIL). As a result, the design doesn’t have to have to compromise in attempting to arrive at a one summary, which can be a recipe for failure.
To recognize how diffusion generative versions operate, it is valuable to explain them based mostly on impression-creating diffusion models. In this article, diffusion designs little by little increase random sound to a 2D impression via a sequence of techniques, destroying the knowledge in the graphic till it becomes almost nothing but grainy static. A neural network is then experienced to get better the original impression by reversing this noising process. The model can then make new facts by starting from a random configuration and iteratively removing the sounds.
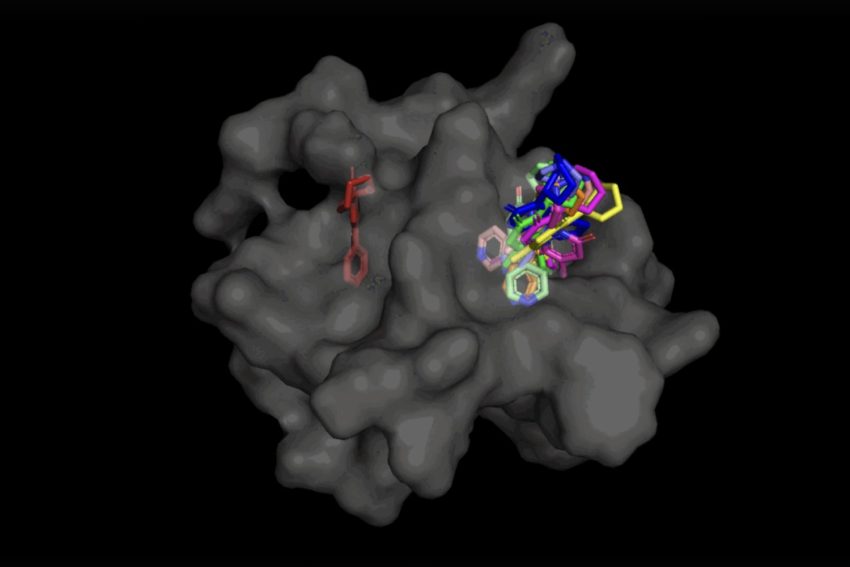
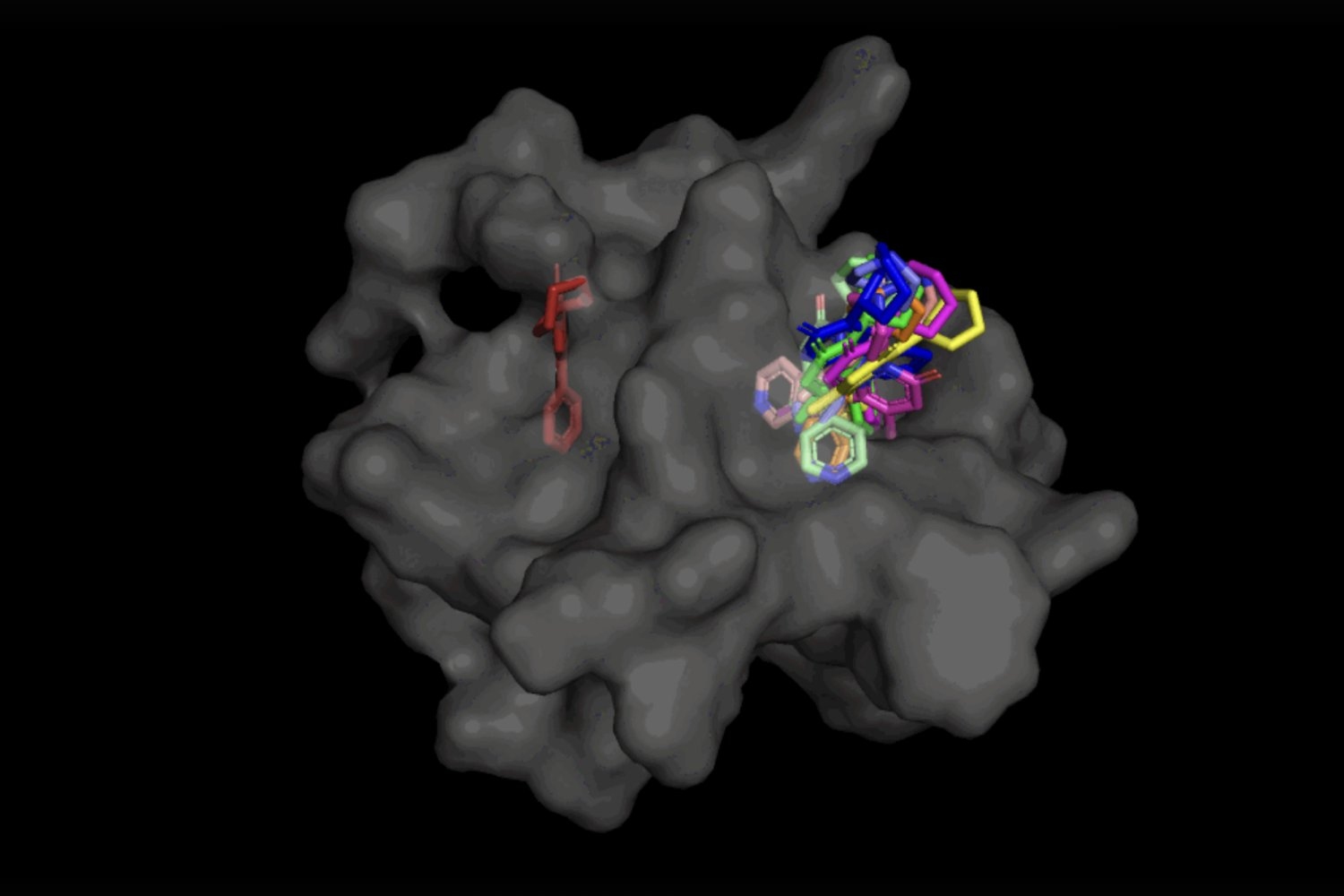
In the circumstance of DiffDock, just after becoming educated on a wide range of ligand and protein poses, the product is able to properly identify numerous binding web pages on proteins that it has in no way encountered before. Instead of generating new image data, it generates new 3D coordinates that aid the ligand come across opportunity angles that would enable it to healthy into the protein pocket.
This “blind docking” strategy results in new options to take benefit of AlphaFold 2 (2020), DeepMind’s famed protein folding AI design. Considering the fact that AlphaFold 1’s original launch in 2018, there has been a great deal of excitement in the research neighborhood around the potential of AlphaFold’s computationally folded protein constructions to support establish new drug mechanisms of action. But point out-of-the-artwork molecular docking equipment have however to display that their performance in binding ligands to computationally predicted constructions is any better than random possibility.
Not only is DiffDock considerably much more precise than earlier approaches to classic docking benchmarks, many thanks to its capability to motive at a higher scale and implicitly design some of the protein versatility, DiffDock maintains superior general performance, even as other docking types begin to are unsuccessful. In the more realistic scenario involving the use of computationally produced unbound protein buildings, DiffDock destinations 22 % of its predictions inside 2 angstroms (extensively viewed as to be the threshold for an exact pose, 1Å corresponds to one particular about 10 billion meters), more than double other docking designs scarcely hovering above 10 p.c for some and dropping as lower as 1.7 %.
These advancements create a new landscape of chances for biological exploration and drug discovery. For occasion, many prescription drugs are identified by using a system acknowledged as phenotypic screening, in which researchers observe the results of a presented drug on a ailment without the need of being aware of which proteins the drug is performing upon. Exploring the system of action of the drug is then crucial to being familiar with how the drug can be enhanced and its likely aspect outcomes. This procedure, acknowledged as “reverse screening,” can be exceptionally demanding and high-priced, but a blend of protein folding approaches and DiffDock could allow accomplishing a huge component of the approach in silico, enabling likely “off-target” facet effects to be recognized early on ahead of clinical trials just take place.
“DiffDock tends to make drug target identification a great deal a lot more achievable. Right before, one particular experienced to do laborious and high priced experiments (months to several years) with each individual protein to outline the drug docking. But now, 1 can display several proteins and do the triaging nearly in a day,” Tim Peterson, an assistant professor at the College of Washington St. Louis School of Medicine, says. Peterson utilized DiffDock to characterize the mechanism of action of a novel drug applicant managing aging-linked illnesses in a the latest paper. “There is a pretty ‘fate enjoys irony’ part that Eroom’s regulation — that drug discovery can take for a longer time and expenses extra income each and every calendar year — is staying solved by its namesake Moore’s law — that computers get faster and less costly each individual 12 months — making use of instruments these types of as DiffDock.”
This operate was done by MIT PhD pupils Gabriele Corso, Hannes Stärk, and Bowen Jing, and their advisors, Professor Regina Barzilay and Professor Tommi Jaakkola, and was supported by the Equipment Discovering for Pharmaceutical Discovery and Synthesis consortium, the Jameel Clinic, the DTRA Discovery of Healthcare Countermeasures Against New and Rising Threats application, the DARPA Accelerated Molecular Discovery plan, the Sanofi Computational Antibody Style and design grant, and a Division of Strength Computational Science Graduate Fellowship.
[ad_2]
Supply hyperlink